comapr
================
[](https://codecov.io/github/ruqianl/comapr/)
[](https://travis-ci.com/ruqianl/comapr)
## Install
``` r
if (!requireNamespace("BiocManager", quietly = TRUE))
install.packages("BiocManager")
BiocManager::install("comapr")
```
## Introduction
<img src='Meta/hexComapr_crop.png' align="right" height="139" />
`comapr` is an R package for finding crossovers for SNP marker intervals
by detecting haplotype shifts across groups of samples.
## From marker genotyping results in data.frame
`comapr` can be applied for detecting crossovers from genotyping results
of makers across samples in a marker by sample data.frame.
``` r
library(comapr)
head(snp_geno)
#> CHR POS rsID C57BL.6J FVB.NJ..i. X92 X93 X94 X95 X96 X97 X98
#> 1 1 4526088 rs13475701 GG CC CC CC CC CC CC CC CG
#> 2 1 5595513 rs13475705 TT CC CC CC CC CC CC CC CT
#> 3 1 6057774 rs3710263 CC TT TT TT TT TT TT TT TC
#> 4 1 6655964 rs13475709 CC GG GG GG GG GG GG GG CG
#> 5 1 21638464 rs6253968 TT CC Fail CC CC CC CC CC Fail
#> 6 1 22665060 rs6361963 AA GG AG GG GG GG GG GG AG
#> X99 X100 X101 X102 X103 X104 X105 X106 X107 X108 X109 X110 X111 X112 X113
#> 1 CG CC CG CG CC CG CG CC CC CC CC CG CC CC CC
#> 2 CT CC CT CT CC CT CT CC CC CC CC CT CC CC CC
#> 3 TC TT TC TC TT TC TC TT TT TT TT TC TT TT TT
#> 4 CG GG CG CG GG CG CG GG GG GG GG CG GG GG GG
#> 5 Fail CC TC TC CC CC CC CC CC CC Fail TC CC CC CC
#> 6 AG GG AG AG GG GG GG AG GG GG GG AG GG GG GG
```
## From sgcocaller’s sparse matrices
`comapr` is also engineered to analyze the outputs from a single-sperm
crossover calling tool
[`sgcocaller`](https://gitlab.svi.edu.au/biocellgen-public/sgcocaller)
``` r
list.files("inst/extdata/")
#> [1] "s1_barcodes.txt" "s1_chr1_altCount.mtx" "s1_chr1_snpAnnot.txt"
#> [4] "s1_chr1_totalCount.mtx" "s1_chr1_vi.mtx" "s1_chr1_viSegInfo.txt"
#> [7] "s1_chr2_altCount.mtx" "s1_chr2_snpAnnot.txt" "s1_chr2_totalCount.mtx"
#> [10] "s1_chr2_vi.mtx" "s1_chr2_viSegInfo.txt" "s1_chr3_altCount.mtx"
#> [13] "s1_chr3_snpAnnot.txt" "s1_chr3_totalCount.mtx" "s1_chr3_vi.mtx"
#> [16] "s1_chr3_viSegInfo.txt" "s1_chr4_altCount.mtx" "s1_chr4_snpAnnot.txt"
#> [19] "s1_chr4_totalCount.mtx" "s1_chr4_vi.mtx" "s1_chr4_viSegInfo.txt"
#> [22] "s1_chr5_altCount.mtx" "s1_chr5_snpAnnot.txt" "s1_chr5_totalCount.mtx"
#> [25] "s1_chr5_vi.mtx" "s1_chr5_viSegInfo.txt" "s2_barcodes.txt"
#> [28] "s2_chr1_snpAnnot.txt" "s2_chr1_vi.mtx" "s2_chr1_viSegInfo.txt"
#> [31] "s2_chr2_snpAnnot.txt" "s2_chr2_vi.mtx" "s2_chr2_viSegInfo.txt"
#> [34] "s2_chr3_snpAnnot.txt" "s2_chr3_vi.mtx" "s2_chr3_viSegInfo.txt"
#> [37] "s2_chr4_snpAnnot.txt" "s2_chr4_vi.mtx" "s2_chr4_viSegInfo.txt"
#> [40] "s2_chr5_snpAnnot.txt" "s2_chr5_vi.mtx" "s2_chr5_viSegInfo.txt"
```
## Visualizing feature of single sperms
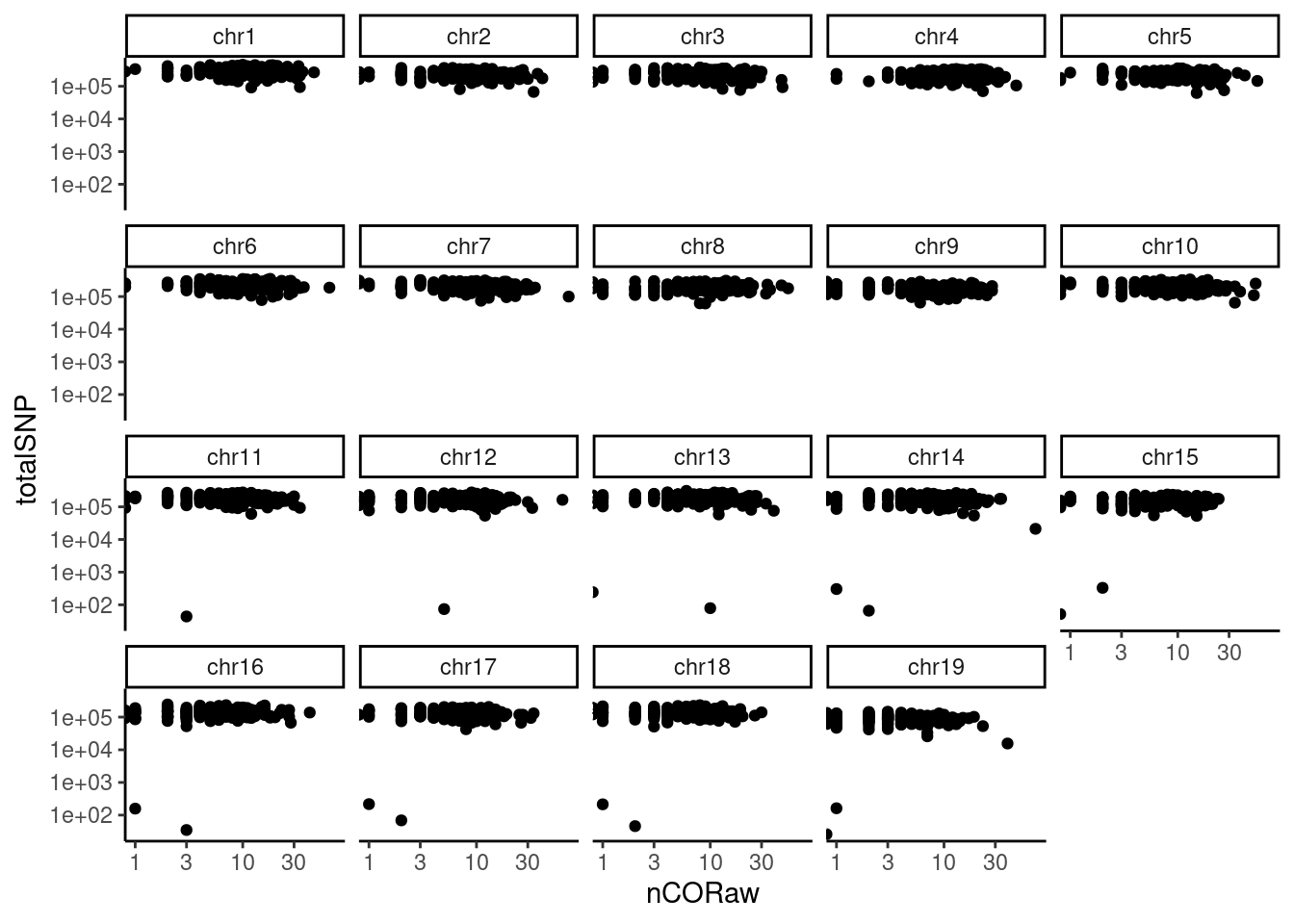
## Crossover positions

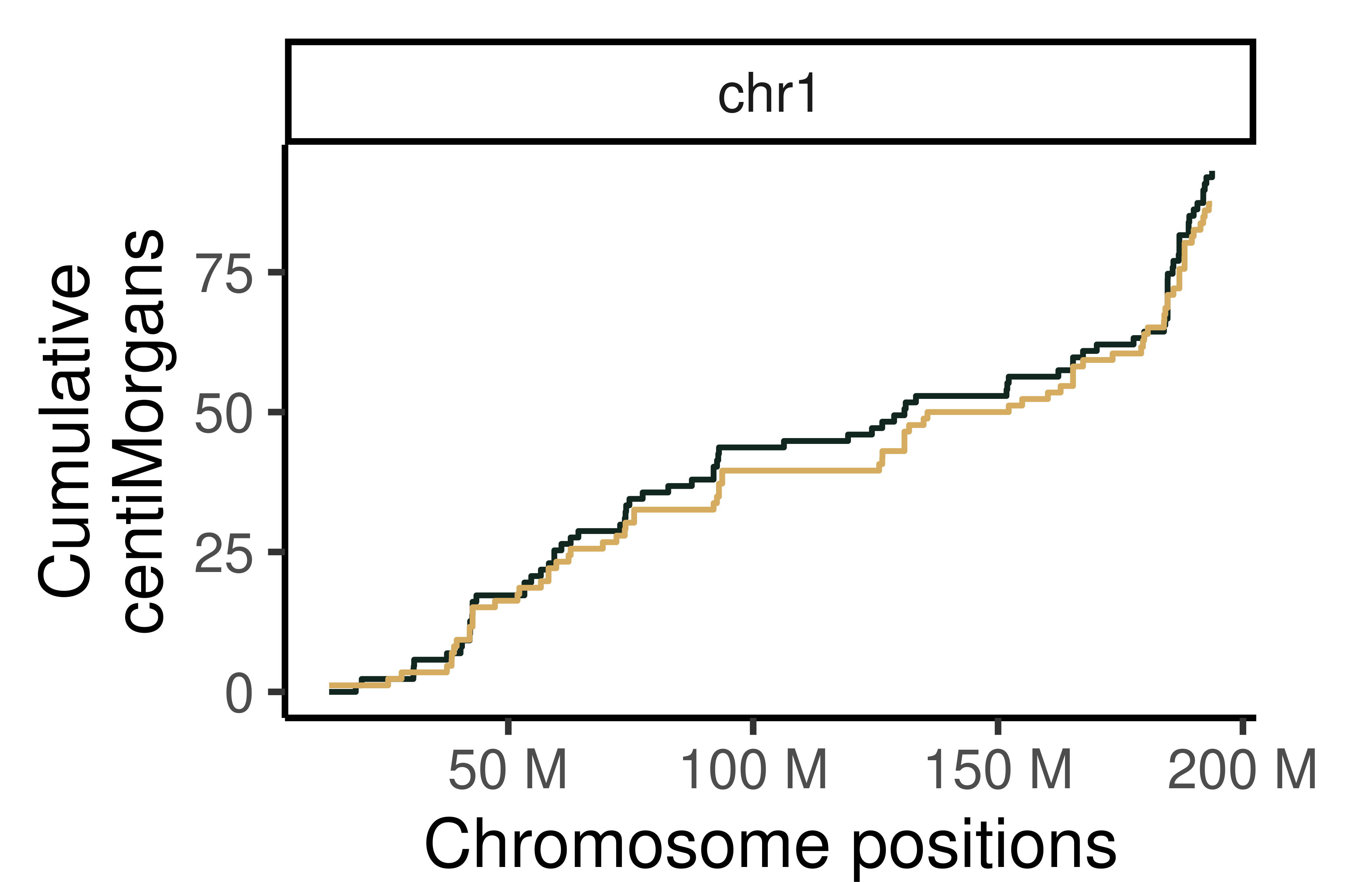
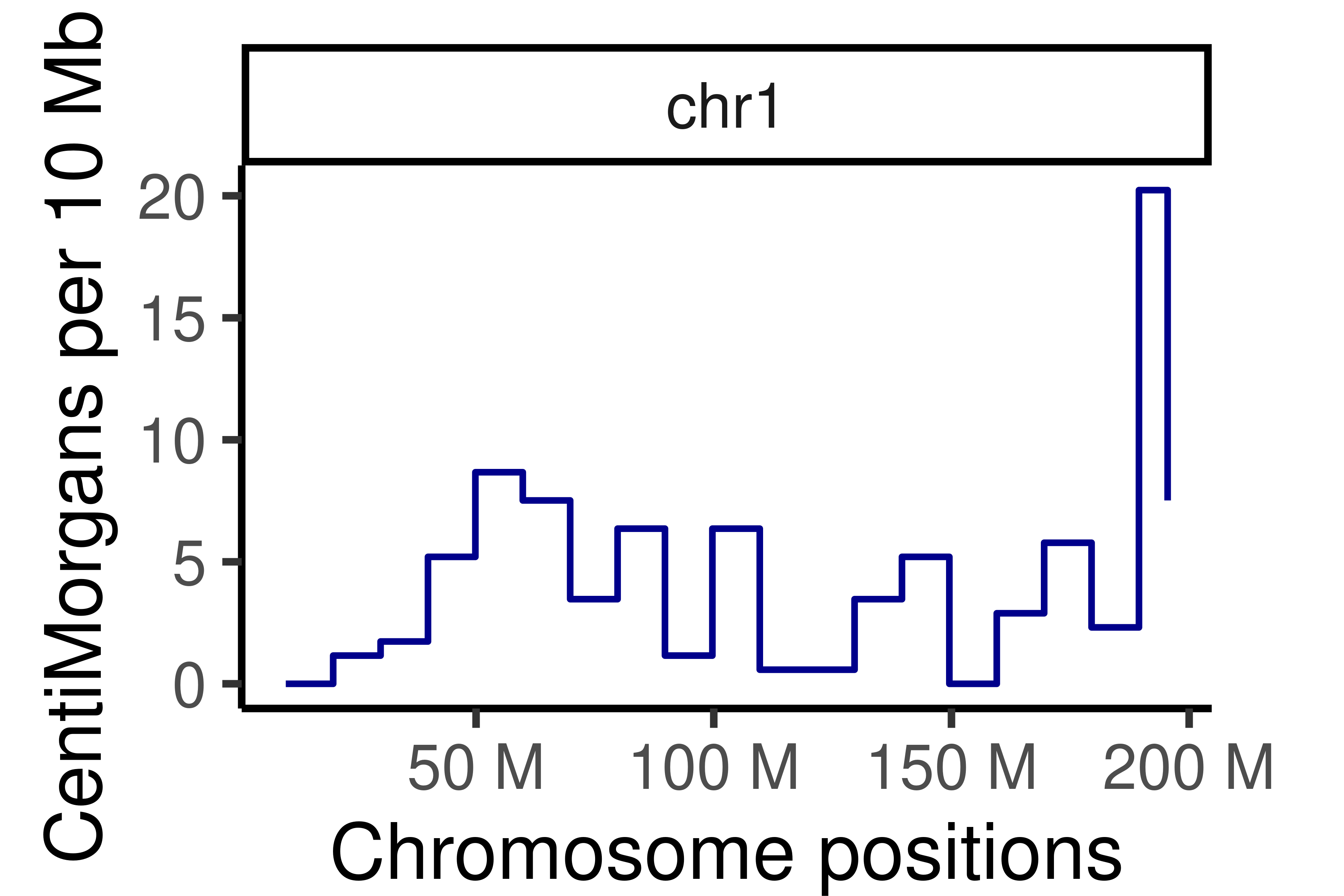
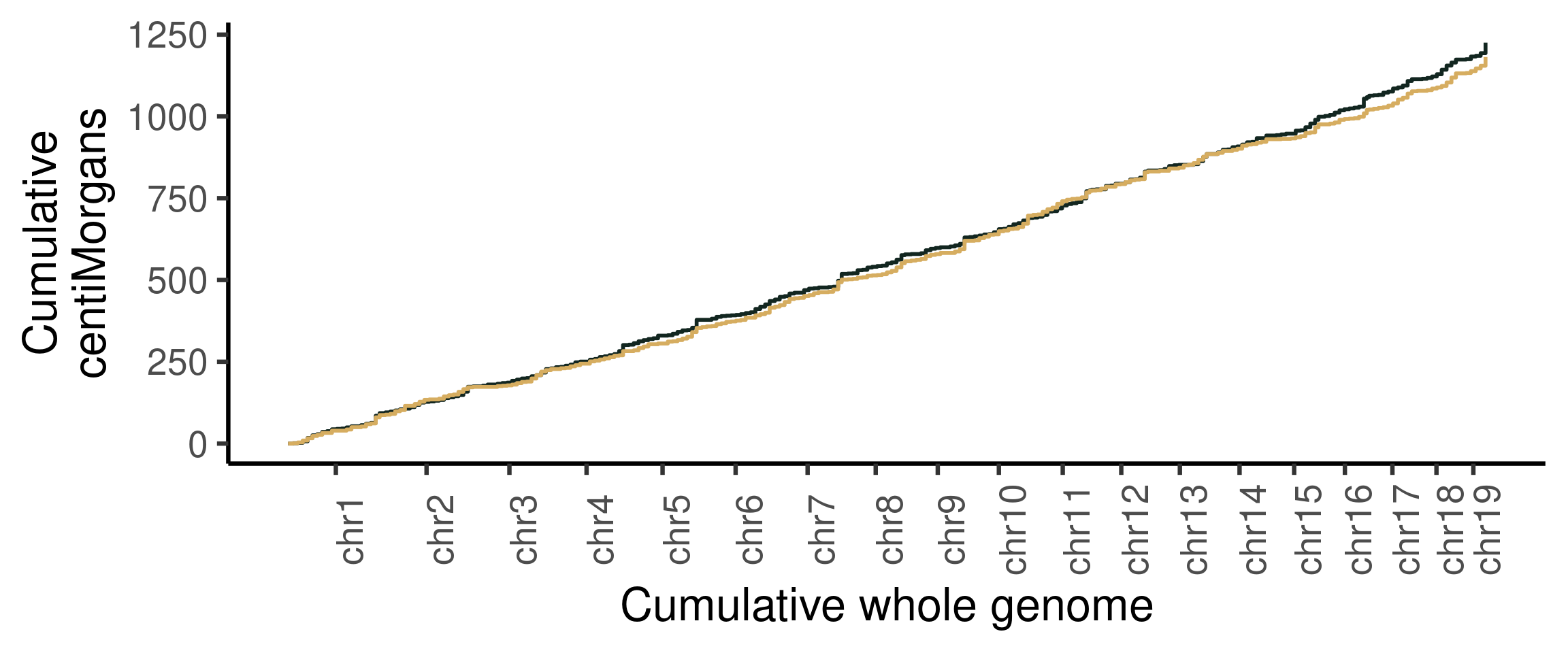
## Cumumative Genetic distances across intervals
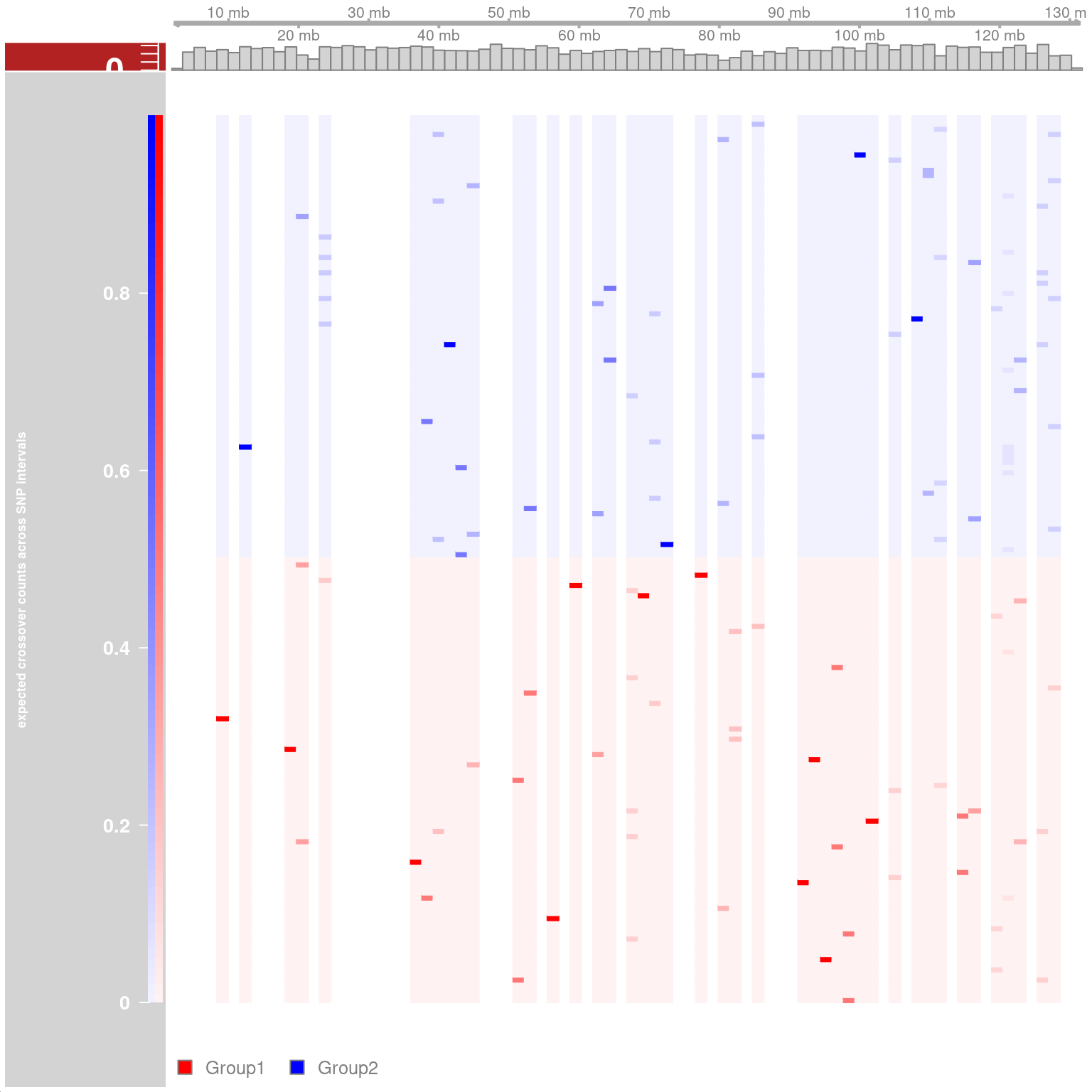
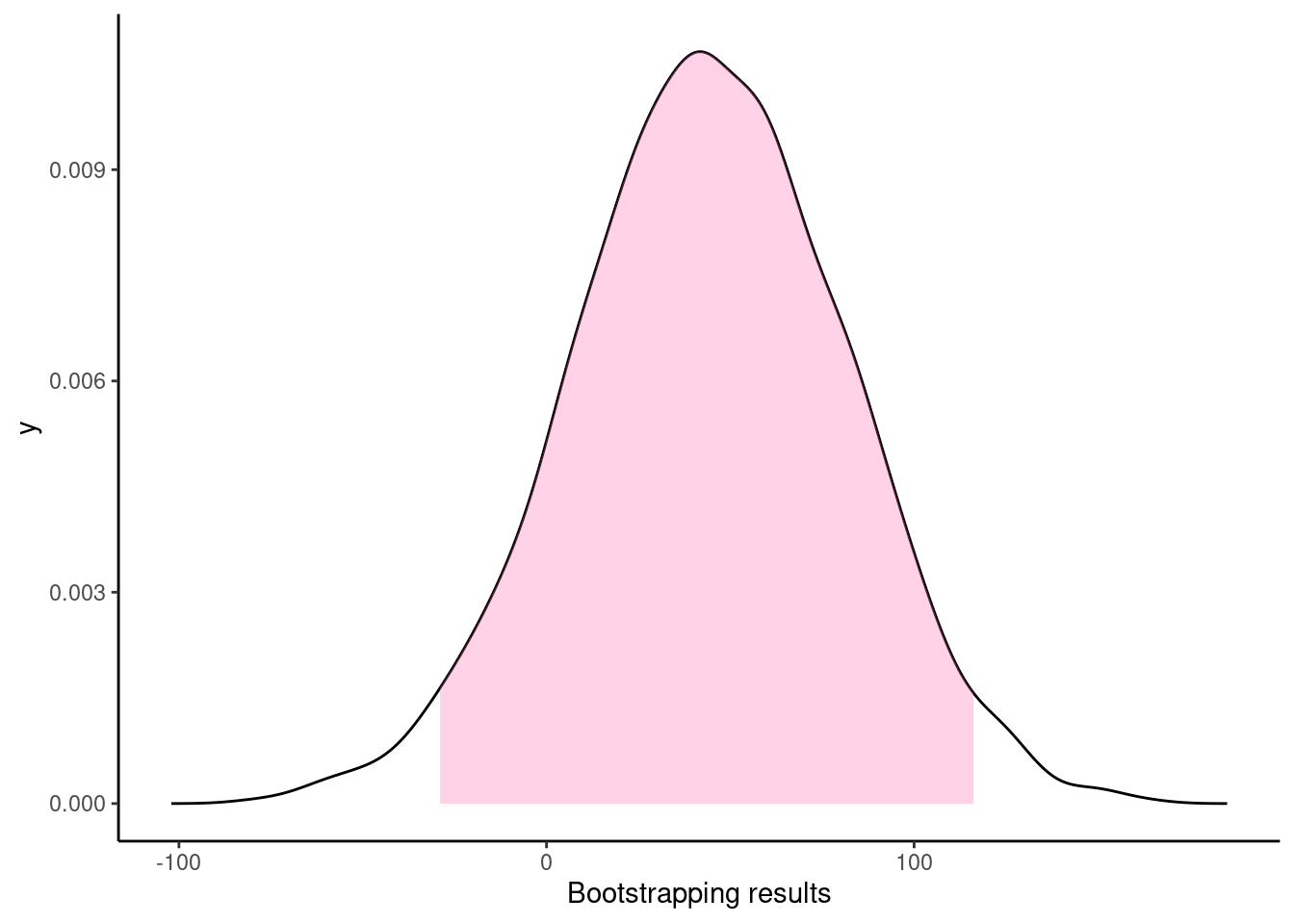
## Resampling methods for comparing groups
## Analysis workflow for demonstration of `comapr` on a single-sperm DNAseq dataset
We demonstrate the usage of `sgcocaller` and `comapr` for identifying
and visualising crossovers regions from single-sperm DNA sequencing
dataset
[here](https://biocellgen-public.svi.edu.au/hinch-single-sperm-DNA-seq-processing/Crossover-identification-with-sscocaller-and-comapr.html)

