<!-- badges: start -->
[](https://www.tidyverse.org/lifecycle/#experimental)
<!--
[](https://bioconductor.org/checkResults/release/bioc-LATEST/Rcollectl)
[](https://bioconductor.org/checkResults/devel/bioc-LATEST/Rcollectl)
-->
[](https://github.com/vjcitn/Rcollectl/actions)
[](https://codecov.io/gh/vjcitn/Rcollectl?branch=main)
<!--
[](https://support.bioconductor.org/t/Rcollectl/)
-->
[](https://github.com/vjcitn/Rcollectl/issues)
<!-- badges: end -->
# Rcollectl: simple interfaces to collectl output
Profiling R computations is supported by Rprof and profvis. These measure memory consumption and
function execution counts and timings. In workflow design we are also interested in CPU load, disk I/O, and network
traffic. There is no standard portable approach to measuring these. In this package, we focus
on linux systems measurement with
the [collectl](http://collectl.sourceforge.net/index.html) suite of tools.
## Notes from the AnVIL workspace description
Our objective in this workspace is demonstration of instrumentation of
bioinformatic tasks. By "instrumentation" we mean the definition and use
of tools that measure computational resource consumption: memory, disk, CPU, network traffic.
(On AnVIL, we do not know how to identify the network device and data on network traffic
are not obtained as of 1/1/2021.)
This Dashboard Description discusses details of an example using [salmon](https://github.com/COMBINE-lab/salmon) to
carry out quantification of an RNA-seq experiment.
## Basic resources
### Workspace backbone
This workspace was initialized using AnVILPublish. The runtime/cloud environment used as of 2020-01-04 is
dockerhub vjcitn/instr:0.0.3
### Raw data
The FASTQ files underlying the "airway" RNA-seq workflow were collected using commands like
```
gsutil -u landmarkanvil2 ls gs://sra-pub-run-3/SRR1039512/SRR1039512.1
gsutil -u landmarkanvil2 cp gs://sra-pub-run-4/SRR1039513/SRR1039513.1 .
gsutil -u landmarkanvil2 cp gs://sra-pub-run-4/SRR1039516/SRR1039516.1 .
gsutil -u landmarkanvil2 cp gs://sra-pub-run-2/SRR1039517/SRR1039517.1 .
gsutil -u landmarkanvil2 cp gs://sra-pub-run-2/SRR1039520/SRR1039520.1 .
gsutil -u landmarkanvil2 cp gs://sra-pub-run-3/SRR1039521/SRR1039521.1 .
```
A public bucket with the extracted fastq was then produced via
```
fastq-dump --split-3 *512.1 &
fastq-dump --split-3 *513.1 &
fastq-dump --split-3 *516.1 &
fastq-dump --split-3 *517.1 &
fastq-dump --split-3 *520.1 &
fastq-dump --split-3 *521.1 &
gzip *fastq
gsutil ls gs://bioc-airway-fastq
gsutil cp SRR1039508.1_2.fastq.gz gs://bioc-airway-fastq/
```
## Quantification process
### Software and indexing resources
Our aim is to analyze the resource consumption in processing this sort of data with
R using Rcollectl. We'll describe how to run salmon to generate quantifications,and
we will attempt to give estimates for processing various volumes of data by various
approaches.
We installed snakemake with pip3 and salmon via
```
wget https://github.com/COMBINE-lab/salmon/releases/download/v1.4.0/salmon-1.4.0_linux_x86_64.tar.gz
```
We obtained GENCODE transcript sequences:
```
wget ftp://ftp.ebi.ac.uk/pub/databases/gencode/Gencode_human/release_36/gencode.v36.transcripts.fa.gz
```
And built the index `gencode.v36_salmon_1.4.0` via
```
salmon index --gencode -t gencode.v36.transcripts.fa.gz -i gencode.v36_salmon_1.4.0
```
### Snakemake for workflow definition
For a single sample (paired-end) we have a snakemake file:
```
rstudio@saturn-1f2f18e5-4182-40c0-8449-1f301b5c3b03:~$ cat snakemake_one
DATASETS = ["SRR1039508.1"]
SALMON = "/home/rstudio/bin/salmon"
rule all:
input: expand("quants/{dataset}/quant.sf", dataset=DATASETS)
rule salmon_quant:
input:
r1 = "fastq/{sample}_1.fastq.gz",
r2 = "fastq/{sample}_2.fastq.gz",
index = "/home/rstudio/gencode.v36_salmon_1.4.0"
output:
"quants/{sample}/quant.sf"
params:
dir = "quants/{sample}"
shell:
"{SALMON} quant -i {input.index} -l A -p 6 --validateMappings \
--gcBias --numGibbsSamples 20 -o {params.dir} \
-1 {input.r1} -2 {input.r2}"
```
When used with
```
snakemake -j1 --snakefile snakemake_one
```
it takes 6 minutes to produce a folder `quants` with content
```
└── SRR1039508.1
├── aux_info
│ ├── ambig_info.tsv
│ ├── bootstrap
│ │ ├── bootstraps.gz
│ │ └── names.tsv.gz
│ ├── expected_bias.gz
│ ├── exp_gc.gz
│ ├── fld.gz
│ ├── meta_info.json
│ ├── observed_bias_3p.gz
│ ├── observed_bias.gz
│ └── obs_gc.gz
├── cmd_info.json
├── lib_format_counts.json
├── libParams
│ └── flenDist.txt
├── logs
│ └── salmon_quant.log
└── quant.sf
```
## Instrumentation of this process
We use the Rcollectl package (which must be installed from github.com/vjcitn/Rcollectl) to
monitor resource consumption while the snakemake process is running.
### Single sample 6 threads
Here is a display of the resource consumption for the single (paired-end) sample:
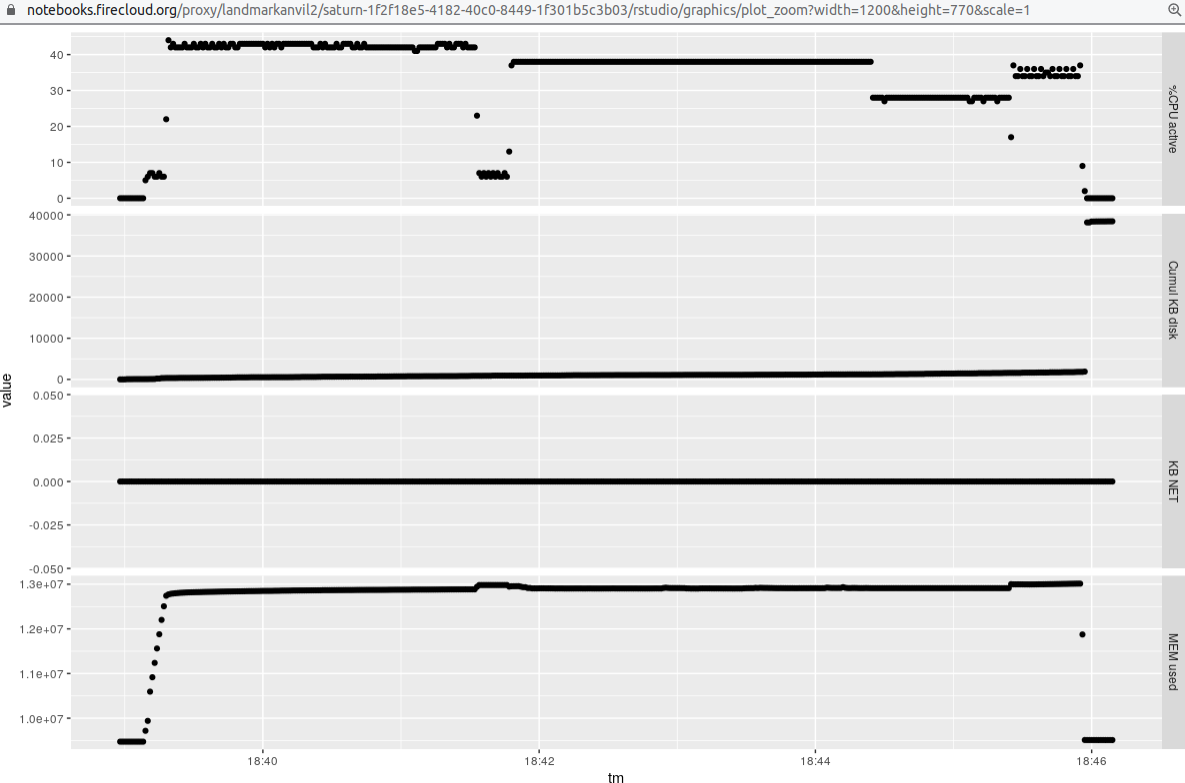
That run uses a setting of `-p 6` for `salmon quant`, which allows the algorithm to use 6 threads.
### Single sample 12 threads
We
used a 16 core machine, and raised the value of `-p` to 12 to obtain the following profile:
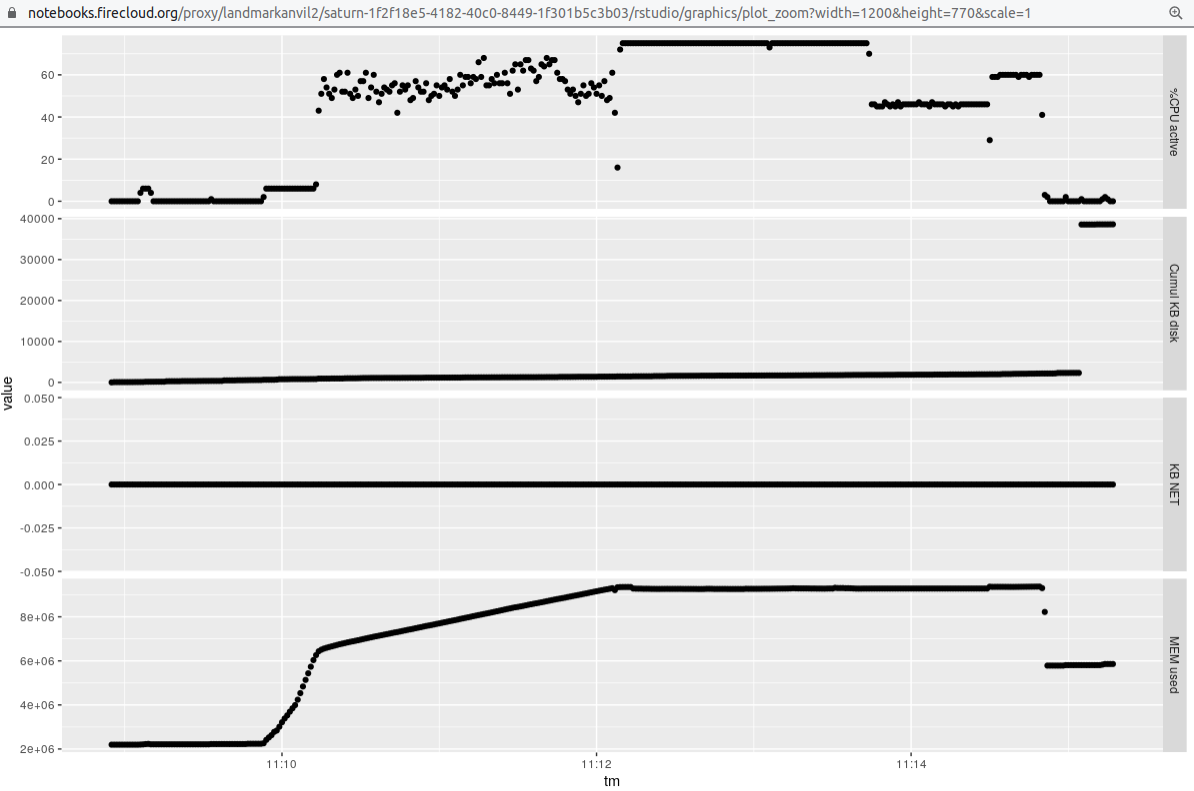
### Eight samples 12 threads
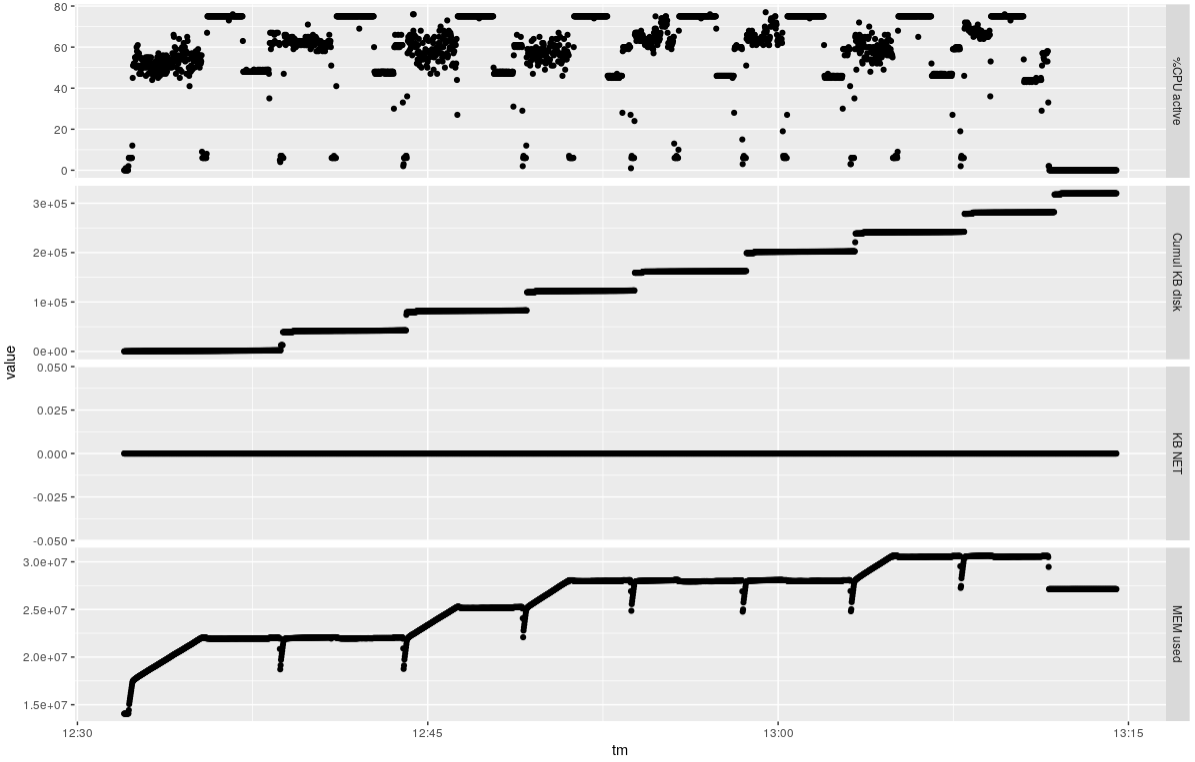
It takes about 40 minutes to do the eight samples, using about 80% of the available CPUs and about 16
GB RAM overall.
### Four samples 3 threads 4 processes (-j4 for snakemake)
Finally, we obtain some data to compare thread-based parallelism to process parallelism. We'll
reduce the number of threads per sample, but run 4 samples at once via snakemake -j argument.
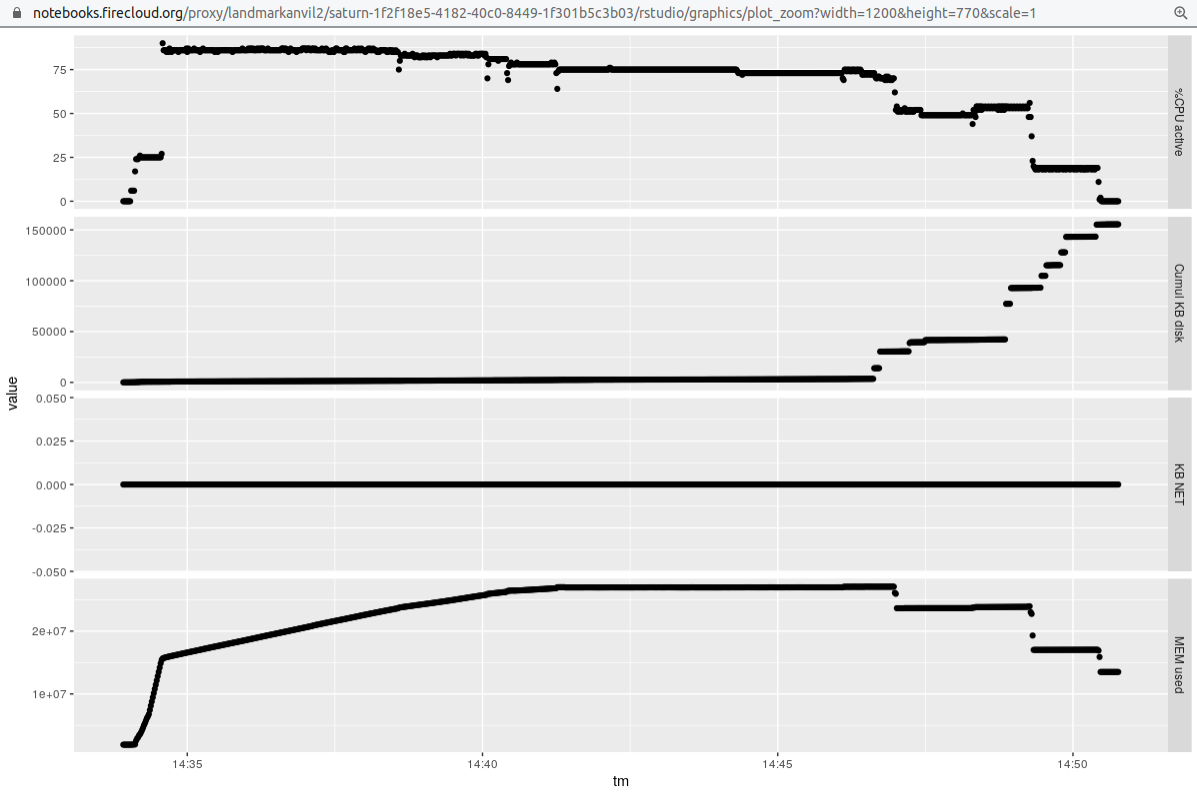
It seems the throughput is a bit better here, but we should check the comparability
of results of the different approaches before declaring this.
## Conclusions
Rcollectl can be used to obtain useful profiling information on AnVIL interactive work. We saw
that the salmon quantification could be tuned to consume 80-90% of available CPUs. The
RAM requirements and disk consumption are measurable. It does not seem possible to decouple
the NCPU from the RAM elements in a fine-grained way in AnVIL interactive cloud environments. For 16 cores we could
choose between 14.4 and 64 GB RAM. 64 GB seems too much for the task we are doing.
Additional work of interest: snakemake is used only because a working snakefile was published. We
would like to be able to conduct these exercises in a more Bioconductor-native way. Specifically,
we should have an object representing the read data in a self-describing way, and use BiocParallel
to manage the execution approach.
Furthermore, as we define the self-describing representation of the read
data in Bioconductor, we should consider how it maps to the AnVIL/Terra-native
structures related to the workspace data tables, and DRS.

