<!-- badges: start -->
[](https://www.nature.com/articles/s41467-024-44761-x)
[](https://www.tidyverse.org/lifecycle/#stable)
[](https://opensource.org/licenses/MIT)
[](https://github.com/js2264/HiCExperiment/actions/workflows/rworkflows.yml)
[](https://js2264.github.io/HiCExperiment)
[](https://js2264.github.io/OHCA/)
<a href=http://bioconductor.org/packages/release/bioc/html/HiCExperiment.html><img alt="Static Badge" src="https://img.shields.io/badge/Bioc_(release)-Landing_page-green?link=http%3A%2F%2Fbioconductor.org%2FcheckResults%2Fdevel%2Fbioc-LATEST%2FHiCExperiment%2F"></a>
<a href=http://bioconductor.org/checkResults/release/bioc-LATEST/HiCExperiment/><img alt="Bioc build (release)" src="https://img.shields.io/badge/dynamic/yaml?url=https%3A%2F%2Fbioconductor.org%2FcheckResults%2Frelease%2Fbioc-LATEST%2FHiCExperiment%2Fraw-results%2Fnebbiolo1%2Fbuildsrc-summary.dcf&query=%24.Status&label=Bioc%20build%20(release)&link=https%3A%2F%2Fbioconductor.org%2FcheckResults%2Frelease%2Fbioc-LATEST%2FHiCExperiment%2F"></a>
<a href=http://bioconductor.org/checkResults/devel/bioc-LATEST/HiCExperiment/><img alt="Bioc build (devel)" src="https://img.shields.io/badge/dynamic/yaml?url=https%3A%2F%2Fbioconductor.org%2FcheckResults%2Fdevel%2Fbioc-LATEST%2FHiCExperiment%2Fraw-results%2Fnebbiolo2%2Fbuildsrc-summary.dcf&query=%24.Status&label=Bioc%20build%20(devel)&link=https%3A%2F%2Fbioconductor.org%2FcheckResults%2Fdevel%2Fbioc-LATEST%2FHiCExperiment%2F"></a>
<!-- badges: end -->
# HiCExperiment
[👉 OHCA book 📖](https://js2264.github.io/OHCA/)
*Please cite:*
Serizay J, Matthey-Doret C, Bignaud A, Baudry L, Koszul R (2024). “Orchestrating chromosome conformation capture analysis with Bioconductor.” _Nature Communications_, **15**, 1-9. [doi:10.1038/s41467-024-44761-x](https://doi.org/10.1038/s41467-024-44761-x).
---
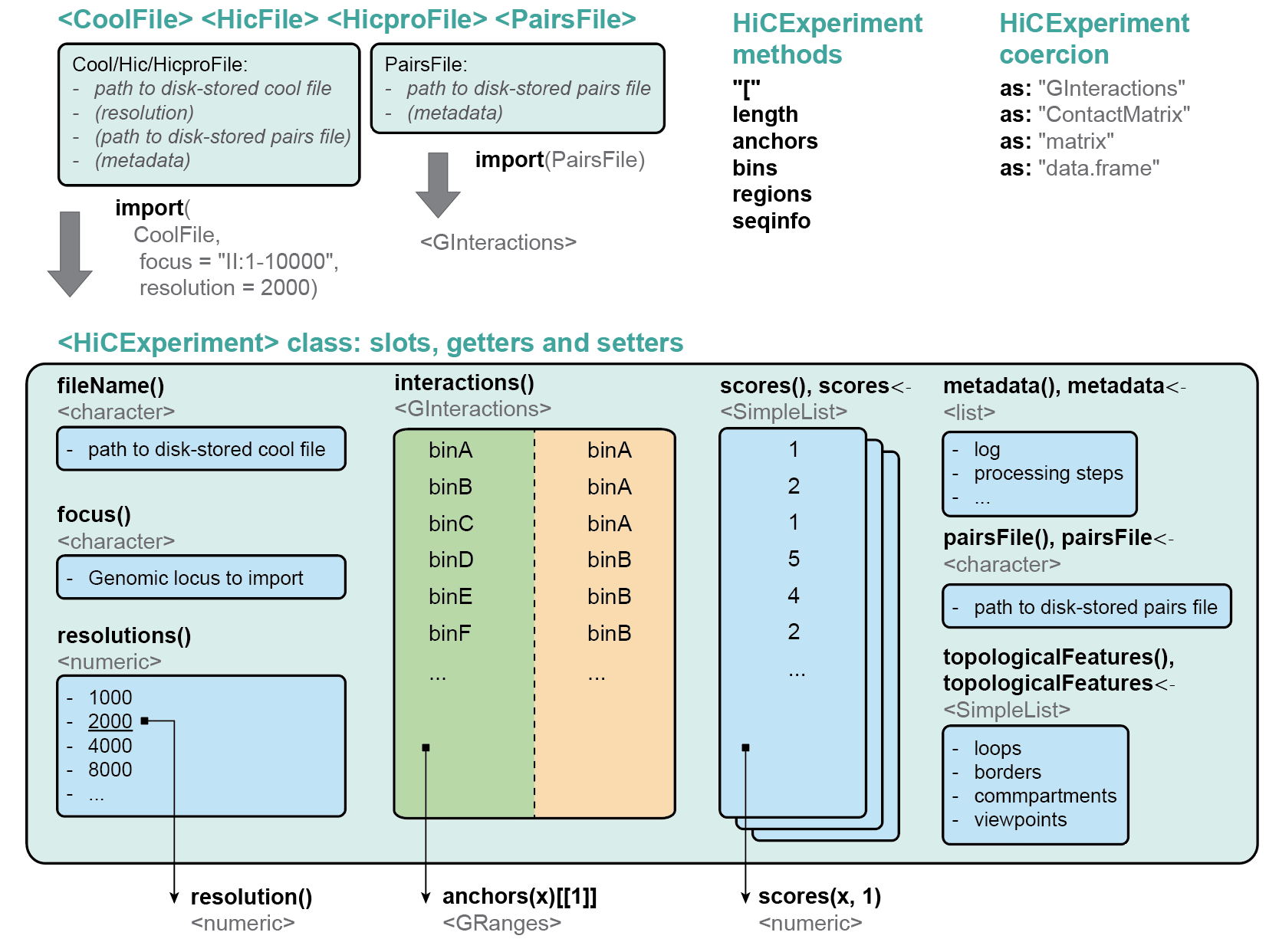
The `HiCExperiment` package provides a unified data structure to import the three main Hi-C matrix file formats (`.(m)cool`, `.hic` and `HiC-Pro` matrices) in R and performs common array operations on them.
The `HiCExperiment` class wraps an (indexed) matrix-like object (i.e. on-disk `.(m)cool`, `.hic` or `HiC-Pro` matrices). For indexed matrices (i.e. `.(m)cool` and `.hic` files), `HiCExperiment` allows one to specfically parse subsets of the contact matrix corresponding to genomic loci of interest, without having to load the entire object in memory.
The `HiCExperiment` package also provides methods to import pairs files generated by `pairtools`/`cooler` workflow, by HiC-Pro pipeline, or any type of tabular pairs format (by indicating the columns containing `chr1`, `start1`, `strand1`, `chr2`, `start2`, `strand2` information).
`HiCExperiment` S4 class is built on pre-existing Bioconductor classes, namely `BiocFile` and
`GInteractions` (Lun, Perry & Ing-Simmons, F1000Research 2016`), and leverages them to
point to on-disk Hi-C matrix files and dynamically parse them into R.
Several other packages rely on the `HiCExperiment` class to provide a rich ecosystem when interacting with Hi-C data.
## Installation
HiCExperiment is an R/Bioconductor package. As such, it can be installed with:
```r
BiocManager::install("HiCExperiment")
```
## Importing a Hi-C matrix file
### `.(m)cool` files:
```r
cool_file <- CoolFile(HiContactsData::HiContactsData('yeast_wt', format = 'cool'))
import(cool_file, focus = "II:10000-100000")
```
```
## `HiCExperiment` object with 3,454 interactions over 90 regions
## -------
## fileName: "/home/rsg/.cache/R/ExperimentHub/36d548fb47bf_7751"
## focus: "II:10,000-100,000"
## resolutions(1): 1000
## current resolution: 1000
## interactions: 3454
## scores(2): count balanced
## topologicalFeatures: loops(0) borders(0) compartments(0) viewpoints(0)
## pairsFile: N/A
## metadata(0):
```
```r
mcool_file <- CoolFile(HiContactsData::HiContactsData('yeast_wt', format = 'mcool'))
import(mcool_file, focus = "II:10000-100000", resolution = 2000)
```
```
## `HiCExperiment` object with 1,004 interactions over 45 regions
## -------
## fileName: "/home/rsg/.cache/R/ExperimentHub/36d590c5583_7752"
## focus: "II:10,000-100,000"
## resolutions(5): 1000 2000 4000 8000 16000
## current resolution: 2000
## interactions: 1004
## scores(2): count balanced
## topologicalFeatures: loops(0) borders(0) compartments(0) viewpoints(0)
## pairsFile: N/A
## metadata(0):
```
### `.hic` files:
```r
hic_file <- HicFile(HiContactsData::HiContactsData('yeast_wt', format = 'hic'))
import(hic_file, focus = "II:10000-100000", resolution = 4000)
```
```
## `HiCExperiment` object with 276 interactions over 23 regions
## -------
## fileName: "/home/rsg/.cache/R/ExperimentHub/7fa45373d163_7836"
## focus: "II:10,000-100,000"
## resolutions(5): 1000 2000 4000 8000 16000
## current resolution: 4000
## interactions: 276
## scores(2): count balanced
## topologicalFeatures: loops(0) borders(0) compartments(0) viewpoints(0)
## pairsFile: N/A
## metadata(0):
```
### HiC-Pro files:
```r
hicpro_file <- HicproFile(
HiContactsData::HiContactsData('yeast_wt', format = 'hicpro_matrix'),
bed = HiContactsData::HiContactsData('yeast_wt', format = 'hicpro_bed')
)
import(hicpro_file)
```
```
## `HiCExperiment` object with 2,686,250 interactions over 11,805 regions
## -------
## fileName: "/home/rsg/.cache/R/ExperimentHub/29210052806_7837"
## focus: "whole genome"
## resolutions(1): 1000
## current resolution: 1000
## interactions: 2686250
## scores(1): counts
## topologicalFeatures: loops(0) borders(0) compartments(0) viewpoints(0)
## pairsFile: N/A
## metadata(1): regions
```
## Importing a pairs file
- `.pairs` files (e.g. from `pairtools` or `cooler`):
```r
pairs_file <- PairsFile(HiContactsData('yeast_wt', format = 'pairs.gz'))
import(pairs_file)
```
```
## GInteractions object with 471364 interactions and 4 metadata columns:
## seqnames1 ranges1 seqnames2 ranges2 | counts frag1 frag2 distance
## <Rle> <IRanges> <Rle> <IRanges> | <integer> <numeric> <numeric> <numeric>
## [1] II 105 --- II 48548 | 1 1358 1681 48443
## [2] II 113 --- II 45003 | 1 1358 1658 44890
## [3] II 119 --- II 687251 | 1 1358 5550 687132
## [4] II 160 --- II 26124 | 1 1358 1510 25964
## [5] II 169 --- II 39052 | 1 1358 1613 38883
## ... ... ... ... ... ... . ... ... ... ...
## [471360] II 808605 --- II 809683 | 1 6316 6320 1078
## [471361] II 808609 --- II 809917 | 1 6316 6324 1308
## [471362] II 808617 --- II 809506 | 1 6316 6319 889
## [471363] II 809447 --- II 809685 | 1 6319 6321 238
## [471364] II 809472 --- II 809675 | 1 6319 6320 203
## -------
## regions: 549331 ranges and 0 metadata columns
## seqinfo: 1 sequence from an unspecified genome; no seqlengths
```
- `.validPairs` files (e.g. from HiC-Pro pipeline):
```r
hicpro_pairs_file <- PairsFile(HiContactsData('yeast_wt', format = 'hicpro_pairs'))
import(hicpro_pairs_file, nrows = 100)
```
```
## GInteractions object with 100 interactions and 4 metadata columns:
## seqnames1 ranges1 seqnames2 ranges2 | counts frag1 frag2 distance
## <Rle> <IRanges> <Rle> <IRanges> | <integer> <numeric> <character> <numeric>
## [1] I 33 --- I 620 | 1 414 HIC_I_1 587
## [2] I 35 --- III 301620 | 1 336 HIC_I_1 NA
## [3] I 41 --- I 68853 | 1 352 HIC_I_1 68812
## [4] I 49 --- I 3233 | 1 311 HIC_I_1 3184
## [5] I 51 --- VIII 197898 | 1 397 HIC_I_1 NA
## ... ... ... ... ... ... . ... ... ... ...
## [96] I 138 --- VIII 326284 | 1 251 HIC_I_1 NA
## [97] I 141 --- I 2466 | 1 231 HIC_I_1 2325
## [98] I 142 --- I 2219 | 1 278 HIC_I_1 2077
## [99] I 142 --- XI 222517 | 1 270 HIC_I_1 NA
## [100] I 142 --- XV 441757 | 1 280 HIC_I_1 NA
## -------
## regions: 158 ranges and 0 metadata columns
## seqinfo: 15 sequences from an unspecified genome; no seqlengths
```
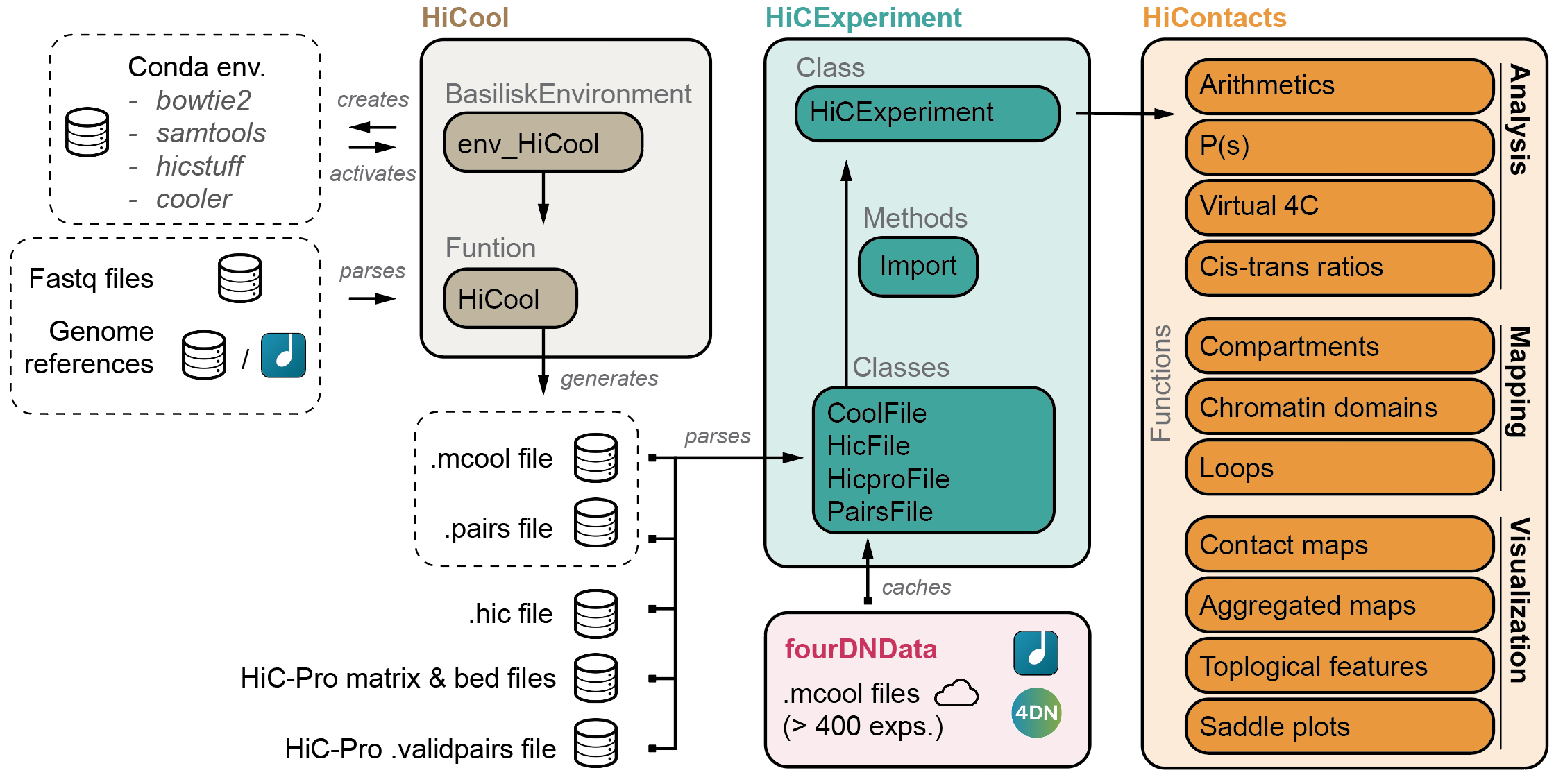
## The `HiCExperiment` ecosystem
### HiContacts
[`HiContacts` package](http://www.bioconductor.org/packages/release/bioc/html/HiContacts.html)
further provides **analytical** and **visualization** tools to investigate Hi-C matrices imported as `HiCExperiment` in R.
Among other features, it provides the end-user with generic functions to annotate topological features in a Hi-C contact map and export them, notably compartments, domains of constrained interactions (so-called TADs) and focal chromatin loops.
### HiCool
`HiCool` package integrates an end-to-end processing workflow, to generate multi-resolution balanced contact matrices from paired-end fastq files of Hi-C experiments.
Under the hood, `HiCool` leverages `hicstuff` and `cooler` to process fastq files into .mcool files. [`hicstuff`](https://github.com/koszullab/hicstuff) takes care of the heavy-lifting, and accurately filters non-informative read pairs out, to retain only informative contacts.
Two important features of `HiCool` are:
1. Its operability within the `R` ecosystem. It relies on `basilisk` to set up a `conda` environment with pinned versions of each software it needs to align, filter and process read pairs into contact matrices.
2. Its transparency. `HiCool` generates QC checks and logs, all embedded in
HTML files to easily inspect the quality of each sample.
### fourDNData
`fourDNData` (read `"4DN Data"`) provides a gateway to
the [4DN data portal](https://data.4dnucleome.org/).
### HiContactsData
[`HiContactsData` package](http://www.bioconductor.org/packages/release/bioc/html/HiContactsData.html)
provides toy datasets to illustrate how the `HiCExperiment` ecosystem works.
## Contributing
We use [devtools](https://github.com/r-lib/devtools) and [testthat](https://github.com/r-lib/testthat) for the development workflow. A Makefile is provided for automation. New functions should be documented with [roxygen2](https://github.com/r-lib/roxygen2) comments and associated tests should be added inside `tests/testthat/`.
* To install the package for development, run `make install`.
* To run tests, run `make test`
* To know more, run `make help`
For development purposes, we provide a DockerHub-hosted `docker` image
with `HiCExperiment` and related packages pre-installed and ready-to-go.
A new image is automatically built on every `push`.
```sh
## To fetch the latest docker image from Docker Hub (for development purposes!)
docker pull js2264/hicexperiment:latest
## To start docker image
docker run -it js2264/hicexperiment:latest /usr/local/bin/R
```
On top of that, for each release, an extra `docker` image is built and
uploaded to the Github Container Repository.
```sh
## To fetch release-specific docker image from Github Container Repo
docker pull ghcr.io/js2264/hicexperiment:0.99.9
## To start docker image
docker run -it ghcr.io/js2264/hicexperiment:0.99.9 /usr/local/bin/R
```

